Síndrome MELAS o encefalomiopatía mitocondrial
El síndrome MELAS (miopatía, encefalopatía, acidosis láctica y episodios semejantes a apoplejías) es un trastorno neurodegenerativo progresivo caracterizado por episodios agudos neurológicos comparables a la apoplejía, asociados a la hiperlactatemia y la miopatía mitocondrial.
Se desconoce la prevalencia exacta de la enfermedad. Los pacientes generalmente se presentan durante la infancia o la edad adulta temprana con crisis agudas, que pueden estar desencadenadas por una infección o por el ejercicio físico. Estas crisis asocian cefaleas con vómitos y, en ocasiones, signos de pseudoapoplejía, como confusión, hemiparesia y hemianopsia.
A menudo se producen en pacientes con síntomas crónicos como debilidad muscular, sordera, diabetes, baja estatura, miocardiopatía, retraso en el desarrollo, pérdidas de memoria o trastornos de atención. La enfermedad está causada por mutaciones en el ADN mitocondrial.
Se han identificado al menos 10 mutaciones diferentes, pero el 80 % de los casos se deben a la mutación 3243A> G en el gen del ARNt de leucina. Así pues, se suele mencionar esta mutación con el nombre de mutación de MELAS a pesar de su asociación con diversos cuadros clínicos: su prevalencia en la población general en Europa se ha estimado en 1/6 250. La mutación 3271T> C del gen ARNt Leu se asocia con el síndrome en un 7,5 % de los pacientes. El diagnóstico del síndrome MELAS se basa en el cuadro clínico y en la resonancia magnética nuclear cerebral.
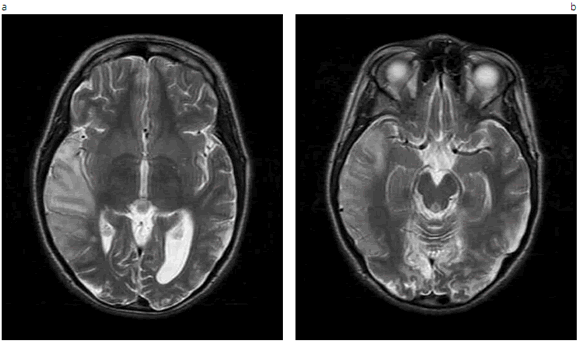
La RM puede revelar muchas lesiones hiperintensas en T2 en sustancia cerebral gris y blanca, mientras que la tomografía computarizada muestra atrofia cerebral y calcificación de los ganglios basales. En ellas se observa que las lesiones no se limitan a los territorios vasculares y por lo tanto que los episodios agudos no son apoplejías típicas. La acumulación anómala de lactato en sangre es frecuente y casi constante en el líquido cefalorraquídeo. La biopsia muscular es anómala en aproximadamente el 85 % de los pacientes. Elanálisis de la actividad de la cadena respiratoria muscular puede revelar una deficiencia del complejo I o una deficiencia combinada de los complejos I y IV.
La identificación de la mutación causal debe tener en cuenta la heteroplasmia constante, es decir, su coexistencia con una población residual de ADN mitocondrial de tipo salvaje. La proporción de las mutaciones puede variar considerablemente entre los tejidos, pero con frecuencia es muy alta y por lo tanto pueden ser investigadas en sangre.
En el síndrome MELAS es muy difícil el asesoramiento genético debido a la heteroplasmia. Las mutaciones en el ADN mitocondrial se transmiten por herencia materna, de modo que un hombre afectado no puede transmitir la enfermedad. La mutación se transmite a lo largo de la línea materna, pero su proporción es esencialmente impredecible.
Aunque proporciones más altas de la mutación en la sangre de la madre tienen como resultado un mayor riesgo de dar a luz un niño con un fenotipo grave, hay muchos ejemplos de segregación extrema de la mutación de la madre al niño, que impiden una orientación genética adecuada genética a nivel individual. La heterogeneidad de las posibilidades en las proporciones de la mutación entre los tejidos dificulta teóricamente el diagnóstico prenatal. Se han realizado muy pocos ensayos clínicos adecuados con pacientes con MELAS.
En un análisis reciente se ha encontrado que el dicloroacetato tiene efectos negativos a medio plazo. La evolución espontánea de la enfermedad con crisis agudas, remisión y recurrencia hace que sea difícil evaluar la mejoría clínica observada en algunos pacientes con MELAS sometidos a tratamientos de apoyo o los efectos perniciosos de tratamientos como el ácido valproico. El pronóstico del síndrome MELAS es desfavorable.
Los pacientes pueden morir durante un episodio de apoplejía y a menudo desarrollan deterioro mental, pérdida de visión y audición y miopatías graves.